BiofacetSNP
BiofacetSNP is a sequence variant data management system.
It takes as input any VCF/gVCF/bcf, and builds up an optimized database allowing deep queries, and incremental merge of set of samples.
It is a two-folds software:
- Technically, in Biofacet Engine 2:
- The provision of a database management layer designed for large scale management of sequence variants.
Using Biofacet's internal Object-Oriented Storage Manager, this layer allows storing, merging and filtering of hundreds of millions of variants of extended genotype values from tens of thousands of samples. - Various algorithmics modules including specific processing of alleles re-alignments producing a proprietary encoding of complex alleles comprising multiple variant events (exhaustive and non-ambiguous representation called the canonical form).
- A sequence variant querying language.
- The provision of a database management layer designed for large scale management of sequence variants.
- A Web Interface (BiofacetSNP Web):
BiofacetSNP Web shares with BiofacetWeb a common architecture and some Web display-layers (see section BiofacetWeb).
BiofacetSNP stores, manages and filters variant sequence data simultaneously at two levels:
- Level 1: variant data (numerical data)
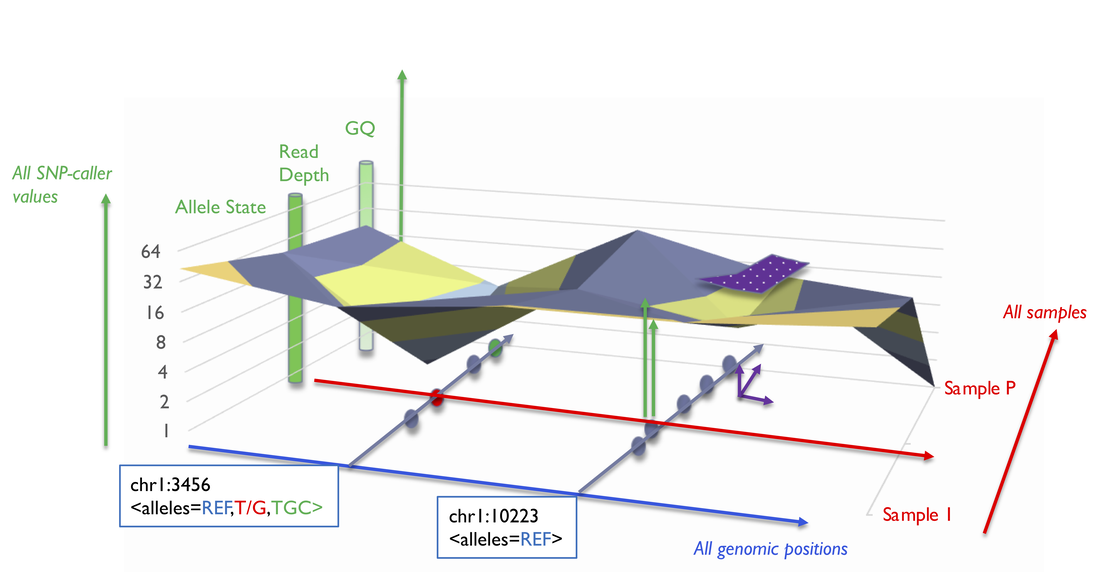
BiofacetSNP storage is a N-dimensional (positions, samples, genotypes) "sparse" matrix where all SNP-caller values from all samples and for all genomic positions are stored. Allele-states including reference calls details are kept and queryable; any incremental merging of new VCF/gVCF samples generates accurate updates of new variants. The purple area is a mapped region from a filter.
- Level 2: textual data Sample Metadata (e.g. phenotypes)
Simultaneously with the numerical data, BiofacetSNP allows to filter sample metadata and/or genome annotation.
BiofacetSNP allows to filter in almost real time loci genome-wide (variants and non-variants), on thousands of (subsets of) samples on multiple criteria. Criteria vary from raw SNP-caller measures (Read Depths, Genotype Quality, …) to any allele-state including indels, with absolute or percentage set thresholds. Filters are dynamic: alleles, allele frequency, and any other property, are recomputed on the fly.
BiofacetSNP allows to express queries such as:
- Loci such as:
- all genotypes from samples set S S={s1,s2,s3, . . ., sN} tagged “Control” be homozygous-ref with Genotype Quality >= Q
- AND at least 80% of calls in "Not Control" samples be heterozygous, with coverage >= 10 in ALT allele
- AND within gene-list {G1,G2, . . ., GK}
- AND that fall in a coding region but not creating a missense event.
- And also, but not limited to:
- Variants polymorphic in a group of samples
- Any variant, or allele state provided: A/G, A/+TGC, … or alleles nucleotide frequency range
- Variants with no flanking variants (N nt) in a group of samples
- Multi-caller support (caller weights / function vote)
Using OpenAPI and Swagger standards, BiofacetSNP can be integrated using:
- Unix command lines
- Python APIs - some are shared with BiofacetWeb
- RESTful Web Services - some are shared with BiofacetWeb
On top of BiofacetSNP, a powerful Web interface is provided.
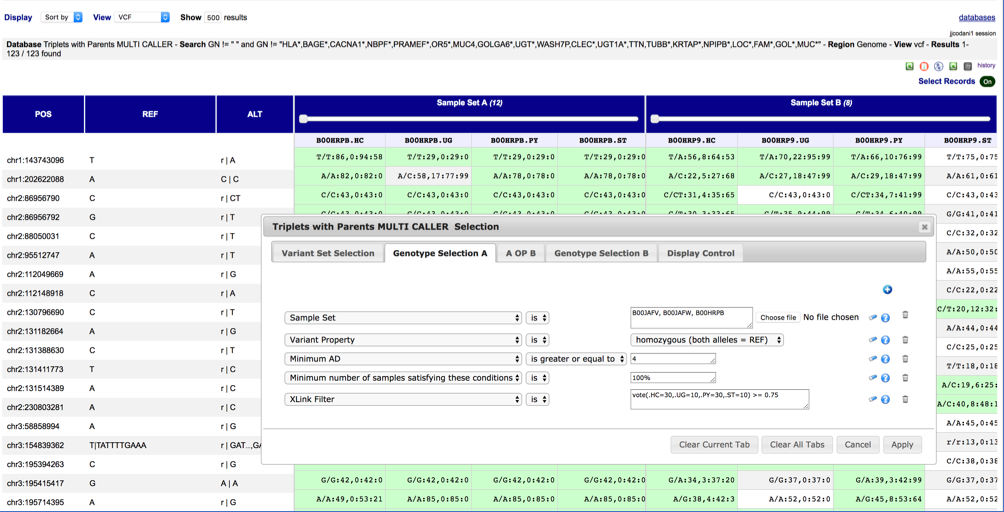
BiofacetSNP Web UI: comparing two datasets of samples (parents/children of a cohort of trios), using a voting function among Haplotype Caller, Platypus, and Samtools SNP callers